Library
Fatty acid oxidation and TG breakdown- Short-answer questions
Question 1: Why are triglycerides a more efficient energy storage form compared to glycogen?
Answer: Triglycerides are highly reduced and stored in a nearly anhydrous form, yielding about 9 kcal/g upon complete oxidation. In contrast, the polar nature of glycogen leads to greater hydration, reducing its energy storage capacity to about 1/6th that of fat.
Question 2: Which hormone plays a crucial role in promoting the breakdown of stored triglycerides into fatty acids and glycerol during hypoglycemic episodes?
Answer: Glucagon, released in response to low blood glucose, predominantly promotes adipolysis.
Question 3: What is the primary location for the beta-oxidation of long-chain fatty acids within the cell?
Answer: The primary location for beta-oxidation is the mitochondrial matrix.
Question 4: How does insulin regulate lipolysis?
Answer: Insulin inhibits lipolysis by reducing cAMP levels. This decrease in cAMP leads to the inactivation of hormone-sensitive lipase, the enzyme responsible for breaking down triglycerides.
Question 5: What is the significance of carnitine in fatty acid oxidation?
Answer: Carnitine acts as a carrier molecule, facilitating the transport of activated long-chain fatty acids across the inner mitochondrial membrane for beta-oxidation.
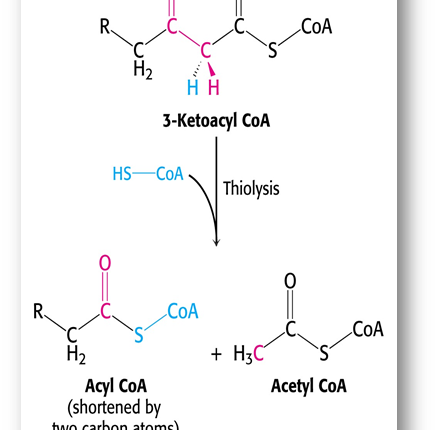
Question 6: What is the role of thiolase in the beta-oxidation spiral?
Answer: Thiolase catalyzes the final step of each beta-oxidation cycle, cleaving the bond between the alpha and beta carbons of the fatty acyl-CoA molecule.
Question 7: How many ATP molecules are generated from the complete oxidation of one molecule of palmitic acid?
Answer: The complete oxidation of one molecule of palmitic acid yields a net of 106 ATP molecules.
Question 8: What are the potential consequences of carnitine deficiency?
Answer: Carnitine deficiency can lead to symptoms such as muscle cramps, weakness, hypoglycemia, and hypoketosis due to the impaired transport of long-chain fatty acids into mitochondria for oxidation.
Question 9: What is the underlying biochemical defect in Jamaican vomiting sickness?
Answer: Jamaican vomiting sickness is caused by the toxin hypoglycin, which inactivates medium and short-chain acyl-CoA dehydrogenases. This inhibition of beta-oxidation leads to hypoglycemia.
Question 10: Besides beta-oxidation, name two other pathways of fatty acid oxidation.
Answer: Two other pathways of fatty acid oxidation are alpha-oxidation and omega-oxidation.
Question 11: What distinguishes alpha-oxidation from beta-oxidation?
Answer: Unlike beta-oxidation, alpha-oxidation involves the removal of a single carbon atom in the form of CO2 per cycle and does not require prior activation of the fatty acid.
Question 12: Which metabolic disorder is associated with the accumulation of phytanic acid due to a deficiency in alpha-oxidation?
Answer: Refsum disease is characterized by the accumulation of phytanic acid due to a deficiency in the enzyme phytanic acid hydroxylase.
Question 13: What is the primary function of omega-oxidation of fatty acids?
Answer: Omega-oxidation is a minor pathway for fatty acid oxidation, but it can become important in conditions like diabetes or starvation, helping eliminate toxic levels of free fatty acids.
Question 14: Where does peroxisomal oxidation of fatty acids occur, and what is its main role?
Answer: Peroxisomal oxidation occurs in peroxisomes and is primarily involved in the shortening of very long-chain fatty acids.
Question 15: What is the genetic basis of Zellweger syndrome, and what are its key clinical features? Answer: Zellweger syndrome is caused by a congenital absence of functional peroxisomes, leading to symptoms like vision disturbances, growth failure, hypotonia, and mental retardation.
Question 16: Name the three main ketone bodies.
Answer: The three main ketone bodies are acetoacetate, beta-hydroxybutyrate, and acetone.
Question 17: In which organ are ketone bodies synthesized, and why are they important?
Answer: Ketone bodies are synthesized in the liver and serve as an alternative fuel source for extrahepatic tissues, particularly during periods of glucose deprivation.
Question 18: Which enzyme is crucial for the utilization of ketone bodies but absent in the liver? Answer: The enzyme succinyl-CoA acetoacetate CoA transferase (thiophorase) is essential for ketone body utilization but is absent in the liver.
Question 19: Explain how the regulation of ketogenesis is linked to lipolysis and the availability of carbohydrates.
Answer: Ketogenesis is upregulated by glucagon, which promotes lipolysis and increases free fatty acid availability. Conversely, insulin inhibits lipolysis and ketogenesis. Carbohydrate deficiency leads to decreased oxaloacetate levels, favoring ketone body formation.
Question 20- List three conditions that can lead to elevated ketone body production (ketosis).
Answer: Three conditions that can cause ketosis are uncontrolled diabetes mellitus, starvation, and chronic alcoholism.
Question 21: A patient presents with severe muscle weakness, hypoglycemia, and an absence of detectable ketone bodies. Biochemical analysis reveals a significant accumulation of long-chain fatty acids within the muscle tissue. What metabolic defect might explain this clinical presentation? Explain the reasoning behind your answer.
Answer: The most likely explanation for these symptoms is carnitine deficiency. Carnitine is a crucial molecule for transporting activated long-chain fatty acids into the mitochondria for beta-oxidation. A deficiency in carnitine would hinder this transport, leading to an accumulation of long-chain fatty acids within the cytoplasm, as observed in the patient. This inability to utilize fatty acids for energy production would explain the muscle weakness. Additionally, the lack of fatty acid oxidation would force the body to rely heavily on glucose for energy, leading to hypoglycemia. The absence of ketone bodies further supports the diagnosis of impaired fatty acid oxidation, as ketone body production is a compensatory mechanism during glucose shortage.
Question 22: Compare and contrast the roles of peroxisomal and mitochondrial beta-oxidation in fatty acid metabolism.
Answer: Both peroxisomal and mitochondrial beta-oxidation contribute to the breakdown of fatty acids, but they differ in their substrate specificity and the end products they generate. Mitochondrial beta-oxidation is the primary pathway for the degradation of short-, medium-, and long-chain fatty acids. It occurs in the mitochondrial matrix and generates FADH2, NADH, and acetyl-CoA, which can enter the TCA cycle for energy production.
Peroxisomal oxidation, on the other hand, primarily targets very long-chain fatty acids (VLCFAs). This process takes place in peroxisomes and differs from mitochondrial beta-oxidation in the initial step. Instead of FADH2, peroxisomal oxidation generates H2O2, which is subsequently detoxified by catalase.
Both pathways ultimately shorten fatty acid chains, but peroxisomal oxidation is crucial for breaking down VLCFAs to a length that can be further processed by mitochondria.
Question 23: How does the regulation of fatty acid oxidation differ between the fed state and the fasting state?
Answer: The regulation of fatty acid oxidation is tightly controlled by hormonal signals and the availability of metabolic intermediates. In the fed state, insulin dominates, promoting glucose uptake and utilization. Insulin indirectly inhibits fatty acid oxidation by stimulating the synthesis of malonyl-CoA, a key intermediate in fatty acid biosynthesis. Malonyl-CoA acts as a potent inhibitor of carnitine palmitoyltransferase I (CPT-I), the enzyme responsible for transporting fatty acids into mitochondria for oxidation.
During fasting, glucagon secretion increases in response to low blood glucose levels. Glucagon stimulates lipolysis in adipose tissue, releasing free fatty acids into circulation. Simultaneously, glucagon inhibits malonyl-CoA synthesis, lifting the inhibition on CPT-I and allowing fatty acid transport into mitochondria for beta-oxidation. This shift in metabolic regulation ensures that fatty acids are used as a primary fuel source during fasting.
Question 24: Why does impairment of beta-oxidation often lead to hypoglycemia, while defects in alpha-oxidation do not?
Answer: The distinct metabolic roles of beta-oxidation and alpha-oxidation explain the difference in their impact on blood glucose levels. Beta-oxidation is the primary pathway for fatty acid catabolism and a major source of energy for most tissues, especially during periods of fasting or low carbohydrate intake. The impairment of beta-oxidation severely limits the body’s ability to derive energy from fatty acids, forcing it to rely heavily on glucose, leading to hypoglycemia.
Alpha-oxidation, in contrast, plays a more specialized role, primarily involved in the breakdown of branched-chain fatty acids like phytanic acid. Defects in alpha-oxidation, as seen in Refsum disease, lead to the accumulation of these specific fatty acids but do not significantly affect overall energy metabolism or glucose homeostasis. Therefore, hypoglycemia is not a typical feature of alpha-oxidation disorders.
Question 25: Explain why the liver is considered the primary site of ketogenesis but cannot utilize ketone bodies for energy production.
Answer: The liver’s unique enzymatic makeup underpins its role in ketone body synthesis and its inability to utilize them. Ketogenesis, the process of ketone body formation from acetyl-CoA, occurs predominantly in the mitochondria of liver cells. The liver expresses high levels of the enzymes HMG-CoA synthase and HMG-CoA lyase, both essential for ketogenesis.
However, the liver lacks a key enzyme required for ketone body utilization: succinyl-CoA acetoacetate CoA transferase (thiophorase). This enzyme is necessary for converting acetoacetate, a primary ketone body, back into acetoacetyl-CoA, which can then be broken down into two molecules of acetyl-CoA to enter the TCA cycle. The absence of thiophorase in the liver prevents the futile cycle of ketone body synthesis and breakdown, ensuring that the ketone bodies produced by the liver are exported and utilized by extrahepatic tissues. This partitioning of metabolic roles allows the liver to serve as a supplier of ketone bodies, which can fuel other organs, especially the brain, during periods of prolonged fasting or glucose shortage.
Question 26- A marathon runner reports early fatigue and muscle cramps during training sessions. Subsequent investigations reveal impaired fatty acid oxidation as a possible cause. Which enzyme deficiency is most likely contributing to this condition?
Answer: Carnitine palmitoyltransferase I (CPT I) deficiency is the enzyme most likely responsible for impaired fatty acid oxidation. This enzyme plays a crucial role in the transport of long-chain fatty acids into the mitochondria for β-oxidation, a key energy-producing process during endurance activities like marathon running.
Question 27: Which enzyme integral to energy metabolism is shared among the metabolic pathways of fatty acid oxidation, ketogenesis, and ketolysis?
Answer: Thiolase is the enzyme common to all three pathways: fatty acid oxidation, ketogenesis, and ketolysis.
- In fatty acid oxidation, thiolase catalyzes the cleavage of acetyl-CoA from acyl-CoA during β-oxidation.
- In ketogenesis, thiolase condenses two molecules of acetyl-CoA to form acetoacetyl-CoA, an early step in ketone body synthesis.
- In ketolysis, thiolase catalyzes the reverse reaction, breaking acetoacetyl-CoA into two acetyl-CoA molecules for energy production.
This multifunctionality makes thiolase integral to energy metabolism.
Question 28: A reduced formation of which metabolic intermediate can upregulate fatty acid oxidation and promote ketosis?
Answer: A reduced formation of malonyl-CoA can upregulate fatty acid oxidation and promote ketosis.
- Malonyl-CoA is a key inhibitor of carnitine palmitoyltransferase I (CPT I), the enzyme responsible for transporting long-chain fatty acids into mitochondria for β-oxidation.
- When malonyl-CoA levels are low, CPT I activity increases, enhancing fatty acid oxidation.
- Increased fatty acid oxidation leads to the production of acetyl-CoA, which drives ketogenesis, resulting in elevated ketone body formation.
Question 29: Which enzyme plays a regulatory role in both fatty acid synthesis and fatty acid oxidation?
Answer: The enzyme Acetyl-CoA Carboxylase (ACC) regulates both fatty acid synthesis and fatty acid oxidation.
- Fatty Acid Synthesis: ACC catalyzes the carboxylation of acetyl-CoA to form malonyl-CoA, which is the first committed step in fatty acid synthesis.
- Fatty Acid Oxidation: Malonyl-CoA, the product of ACC, inhibits carnitine palmitoyltransferase I (CPT I), the enzyme responsible for transporting fatty acids into mitochondria for β-oxidation.
By controlling malonyl-CoA levels, ACC plays a central role in determining whether the cell favors fatty acid synthesis or oxidation, depending on energy demands and metabolic state.
Question 30: Explain the biochemical basis of ketoacidosis in starvation and diabetes mellitus.
Answer: Ketoacidosis arises from an excessive accumulation of ketone bodies in the blood, leading to a decrease in blood pH (acidosis).
Starvation: During prolonged fasting or starvation, the body’s glucose reserves are depleted. To compensate, the body increases lipolysis – the breakdown of stored triglycerides in adipose tissue into free fatty acids and glycerol. These free fatty acids are transported to the liver, where they undergo beta-oxidation, producing acetyl-CoA. Normally, acetyl-CoA enters the TCA cycle for energy production. However, in starvation, oxaloacetate, a key intermediate in the TCA cycle, becomes depleted due to limited glucose availability. This shortage of oxaloacetate forces acetyl-CoA to be shunted towards ketogenesis, leading to an overproduction of ketone bodies (acetoacetate, beta-hydroxybutyrate, and acetone). The liver releases these ketone bodies into the bloodstream, where they can be utilized by extrahepatic tissues, such as the brain and heart, as an alternative fuel source.
Diabetes Mellitus: In type 1 diabetes mellitus, the pancreas produces little or no insulin. Insulin is a crucial hormone that regulates glucose uptake and metabolism. Without sufficient insulin, Glucose cannot enter cells efficiently, leading to hyperglycemia (high blood glucose). Cells are essentially starved of glucose despite its abundance in the blood. Similar to starvation, this glucose deprivation triggers lipolysis and increases the flux of free fatty acids to the liver. The lack of insulin also disrupts the normal regulation of ketogenesis, leading to uncontrolled ketone body production. The accumulation of ketone bodies, specifically acetoacetate and beta-hydroxybutyrate, exceeds the body’s capacity to utilize them, leading to ketoacidosis.
Clinical Implications of Ketoacidosis: The excessive accumulation of ketone bodies in both starvation and uncontrolled diabetes has several serious consequences:
Metabolic Acidosis: Acetoacetate and beta-hydroxybutyrate are acidic. Their buildup in the blood lowers the pH, leading to metabolic acidosis. This can disrupt various physiological processes and impair organ function.
- Dehydration: The kidneys attempt to excrete excess ketone bodies in the urine, leading to increased urination and fluid loss, contributing to dehydration.
- Electrolyte Imbalance: Ketoacidosis can disrupt electrolyte balance, particularly potassium and sodium levels, further compromising cellular function.
- Coma and Death: If left untreated, severe ketoacidosis can lead to coma and even death.
Distinguishing Ketoacidosis in Starvation vs. Diabetes: While the underlying metabolic pathway is similar in both conditions, there is a key difference:
- Starvation: Ketogenesis in starvation is a regulated response to conserve glucose for vital organs like the brain. The level of ketosis is generally moderate, and ketoacidosis is less likely to develop.
- Diabetes: In uncontrolled diabetes, ketogenesis is dysregulated due to insulin deficiency. This can lead to a rapid and severe buildup of ketone bodies, making ketoacidosis a serious and potentially life-threatening complication.