Case details
A 63-year-old female presented to the clinic with recurrent mid-epigastric pain over the last 3 months. She also reported feeling tired and had to increase the amount of ibuprofen needed for the relief of her arthritis. She reported some relief of epigastric pain shortly after eating, but then the discomfort returned. She had tried various over-the-counter medications without relief. She denied nausea, vomiting, and diarrhea.
On examination, she was pale with no jaundice, and her pulse and blood pressure were normal. She was found to have mild mid-epigastric tenderness. A CBC revealed normal white blood cell count and microcytic anemia consistent with iron deficiency. The patient was referred to a gastroenterologist who performed an upper GI endoscopy that identified gastric ulcers. He stated that he suspected that ibuprofen, a nonsteroidal anti-inflammatory drug (NSAID), was the causative agent and suggested switching from ibuprofen to coxibs, such as celecoxib.
What is the likely biochemical etiology of the disorder?
Why NSAID was changed to Celecoxib?
Case details
The NSAIDs include a wide variety of medications, including aspirin, ibuprofen, naproxen, and indomethacin. These medications are used for pain, inflammation, dysmenorrhea, headache, arthritis, or fever. These compounds act as anti-inflammatory and antipyretic agents, inhibiting COX catalysis by prostaglandin H synthase (PGHS). Side effects and complications due to NSAIDs are considered the most common drug-related toxicities in the United States. The spectrum of NSAID-induced morbidity ranges from nausea and dyspepsia (prevalence reported as high as 50–60%) to a serious gastrointestinal complication such as endoscopy-documented peptic ulceration (15–30% of individuals taking NSAIDs regularly) complicated by bleeding or perforation in as many as 1.5% of users per year. About 20,000 patients die each year from serious gastrointestinal complications from NSAIDs.
Pathophysiology
Prostaglandins play a critical role in maintaining gastroduodenal mucosal integrity and repair. Therefore, interruption of prostaglandin synthesis can impair mucosal defense and repair, thus facilitating mucosal injury via a systemic mechanism. Injury to the mucosa also occurs as a result of the topical encounter with NSAIDs. Aspirin and many NSAIDs are weak acids that remain in a nonionized lipophylic form when found within the acid environment of the stomach. Under these conditions, NSAIDs migrate across lipid membranes of epithelial cells, leading to cell injury once trapped intracellularly in an ionized form. Topical NSAIDs can also alter the surface mucous layer, permitting back diffusion of H+ and pepsin, leading to further epithelial cell damage. Moreover, enteric-coated or buffered preparations are also associated with the risk of peptic ulceration.
Basic concept
Prostanoids Synthesis (figure-1)
The prostanoid signaling cascade begins with an external stimulus, most often the binding of a ligand to a cell surface receptor that activates one or more phospholipase A2. The latter are enzymes that release Arachidonic acid from its esterified form in membrane phospholipids such as phosphatidylethanolamine and phosphatidyl Inositol.
Arachidonate is converted to PGH2 by one of the isoforms of PGH synthase (PGHS-1 or -2), enzymes localized to the endoplasmic reticulum membrane and the nuclear envelope.
PGH2 is, in turn, metabolized to the prostanoid lipid signals (PGD2, PGE2, PGF2α, PGH2, PGI2, or TXA2) by one of the secondary enzymes that are named for the individual prostanoid produced. The type of prostanoid produced is determined by which downstream enzyme is present; usually, one downstream enzyme predominates in a given cell. For example, the prominent secondary enzyme in platelets is thromboxane synthase, whereas vascular endothelial cells feature prostacyclin (PGI) synthase.
Conversion of Arachidonate to PGH2 is a key regulatory step in prostanoid biosynthesis.
Each PGHS isoform catalyzes two separate reactions.
The first reaction (arachidonate→PGG2) involves the insertion of two oxygen molecules and cyclization of the fatty acid backbone. This step is catalyzed by the cyclo-oxygenase activity of PGHS-1 or -2; it is these cyclo-oxygenase activities (also called COX-1 and COX-2) that are inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs).
The second step (PGG2→PGH2) involves the reduction of the hydroperoxide on C15 to alcohol and is catalyzed by the peroxidase activity of PGHS-1 or -2.
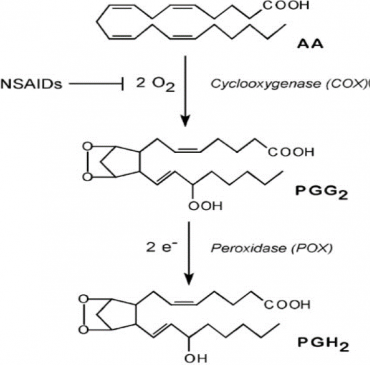
Figure 1- showing the steps of Prostanoid synthesis
COX-1 and COX2
Although both PGHS isoforms have cyclooxygenase and peroxidase activities and are structurally similar proteins, they have very distinct pathophysiologic functions (Figure 2).
COX-1- Many cells, including platelets and gastric mucosal cells, have moderate levels of the “basal” isoform, PGHS-1. Functions attributed to PGHS-1 include regulating hemostasis and vascular tone, renal function, and maintaining gastric mucosal integrity.
COX-2- A smaller number of cells, such as macrophages, vascular endothelial cells, and fibroblasts, dramatically upregulate levels of the “inducible” isoform, PGHS-2, in response to cytokines or mitogens. PGHS-2 has been implicated in cell proliferation, inflammation, carcinogenesis, and parturition.
Cyclo-oxygenase inhibitors– Many cyclooxygenase inhibitors have been developed.
Aspirin was one of the earliest NSAIDs discovered and is now widely used as an analgesic and anti-inflammatory agent. More recently, aspirin has emerged as a very useful antithrombotic agent because of its action against platelet cyclooxygenase activity. Aspirin brings about inhibition of the enzyme by covalent modification making the transition irreversible.
Flurbiprofen and indomethacin are able to inhibit both PGHS-1 and -2, although they do not covalently modify either protein. Ibuprofen forms only transient complexes with both PGHS isoforms.
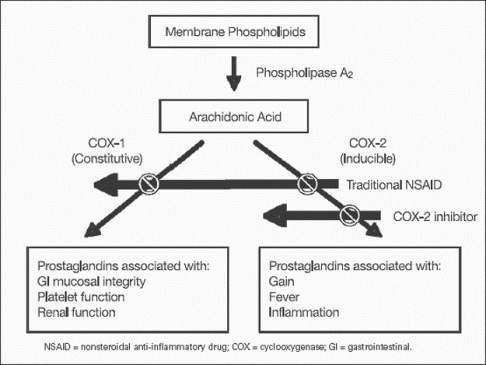
Figure 2– showing the mechanism of action of traditional v/s selective COX inhibitors
Selective COX-2 inhibitors
The recently developed coxibs (such as celecoxib and rofecoxib) derive their exquisitely selective inhibition of PGHS-2 cyclooxygenase from their ability to bring about a noncovalent modification of PGHS-2 and not of PGHS-1. This selectivity has made the coxibs very useful for anti-inflammatory and antiproliferative therapy with reduced gastrointestinal side effects, but it also makes them ineffective as antiplatelet agents and consequently can increase cardiovascular risks.
Low-dose aspirin is often used in antithrombotic prophylaxis
Thromboxane produced by platelet COX-1 in concert with a downstream enzyme is prothrombotic, so aspirin and other NSAIDs cause platelet dysfunction and increase bleeding time. Aspirin is unusual in that it causes covalent, irreversible inhibition of the COX protein, whereas other NSAIDs have noncovalent, reversible actions. Thus, platelets, because they cannot synthesize more COX protein, are irreversibly affected by aspirin but only temporarily affected by other NSAIDs. Due to this reason only Low-dose aspirin is often used in antithrombotic prophylaxis as in the prevention of stroke, I.H.D.etc.
The prothrombotic and vasoconstrictive actions of COX-1-derived Thromboxane in the vasculature are opposed by an antithrombotic and vasodilative prostaglandin, prostacyclin, that originates from COX-2 in vascular endothelial cells. The COX-2 selective coxibs thus tend to decrease prostacyclin levels in the vasculature without reducing the thromboxane levels. This tendency is thought to explain the small but significant increase in cardiovascular risk that recently led to the withdrawal of two coxibs from the U.S. market. Moreover, COX proteins are irreversibly affected by aspirin but only temporarily affected by other NSAIDs.