Case details
A severe form of obstructive lung disease, starting with dyspnea and leading progressively toward emphysema, was found in several members of the family. One brother had died earlier of lung disease.
Blood plasma from his surviving brother and sister showed an abnormally low concentration of α1-Antitrypsin (5.5 μm, normal is 20–48 μm), the α1-plasma fraction also moved abnormally on isoelectric focusing gel electrophoresis.
What is the relationship of emphysema with α1-Antitrypsin?
What is the cause of abnormal movement during electrophoresis?
What are the prospects for the treatment of this emphysema?
Case discussion
Emphysema is a lung disease characterized by the destruction of the alveolar walls. Its causes are complex but airway infection, cigarette smoking, air pollution, and familial factors are involved. A deficiency in plasma α1- Antitrypsin leads to the development of emphysema and less frequently hepatic disease.
α1-Antitrypsin
It was so named because it is a good inhibitor in vitro of pancreatic protease trypsin. It is synthesized primarily in the liver and is secreted into the bloodstream. The inhibitor makes-up most of the proteins in the α1-globulin band following the electrophoresis of plasma.
Pathogenesis of α1-Antitrypsin deficiency
In healthy individuals, the lungs contain a natural enzyme called neutrophil elastase that, under normal circumstances, helps the lungs by digesting damaged, aging cells and bacteria. This process promotes the healing of lung tissue. Unfortunately, this enzyme is nonspecific in nature and eventually attacks the lung tissue instead of helping it heal. That is where alpha antitrypsin (AAT) comes into action, by destroying the enzyme before it can cause actual damage to healthy lung tissue. It promotes the lungs to function normally. When there is not enough AAT, however, the lung tissue will continue to be destroyed, which may lead to chronic lung disease, emphysema.
Inheritance
The two patients described in this case are male and female, suggesting that the deficiency is inherited as an autosomal trait. Genetics has been studied in a large number of patients and family members. Analysis suggests that two independent alleles exist on chromosome 14. The deficiency is inherited as a codominant trait; that is, both alleles contribute to the disease and neither is dominant over the other. The two genes that each person carries for α1-Antitrypsin may be diverse, more than 100 variants are known.
The two common deficiency variants of a1-antitrypsin, S, and Z, result from point mutations in the α1- antitrypsin gene and are named on the basis of their slower electrophoretic mobility on isoelectric focusing analysis compared with the normal M allele. S α1-antitrypsin (264Glu→Val) is found in up to 28 percent of Southern Europeans and, although it results in plasma α1-antitrypsin levels that are 60 percent of the M allele, it is not associated with any pulmonary sequelae. This is usually sufficient to protect the lungs from the effects of elastase in people who do not smoke.
The Z variant (342Glu→Lys) results in a more severe deficiency that is characterized, in the homozygote, by plasma α1-antitrypsin less than 15 percent of normal, and patients are likely to develop panacinar emphysema at a young age; 50 percent of these patients will develop liver cirrhosis because the A1AT is not secreted properly and instead accumulates in the liver.
Clinical Manifestations
The first signs and symptoms of lung disease caused by alpha-1 antitrypsin deficiency usually appear between the ages of 20 and 50. The earliest symptoms are:
- Shortness of breath following mild activity,
- Reduced ability to exercise and
- Wheezing.
Other signs and symptoms can include:
- Unintentional weight loss,
- Recurring respiratory infections,
- Fatigue,
- Rapid heartbeat upon standing.
Advanced lung disease leads to emphysema, in which small air sacs in the lungs (alveoli) are damaged.
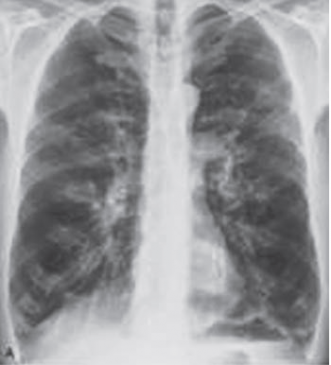
Figure 1: Hyperinflation due to panacinar emphysema. Posteroanterior chest radiograph shows that the dome of the right hemidiaphragm is below the level of the anterior right 7th rib, consistent with increased lung volumes. Also noted is a decrease in the peripheral vascular markings. The patient had severe panacinar emphysema due to alpha-1 antitrypsin deficiency.
Characteristic features of emphysema include:
- Difficulty breathing,
- A hacking cough, and
- A barrel-shaped chest.
Smoking or exposure to tobacco smoke accelerates the appearance of symptoms and damage to the lungs.
About 10 percent of infants and 15 percent of adults with alpha-1 antitrypsin deficiency have liver damage.
Signs of liver disease can include:
- Ascites,
- Edema feet or legs, and
- Jaundice.
Diagnosis
The following tests may also help with diagnosis:
- Alpha-1 antitrypsin estimation
- Arterial blood gases
- Chest X-ray (Figure 1)
- CT scan of the chest
- Genetic testing
- Pulmonary function tests.
Measurement of alpha-1 antitrypsin: Isoelectric focusing on polyacrylamide gel is useful in distinguishing specific variants to establish the specific phenotype.
Effect of smoking on antitrypsin levels: Smoking greatly exacerbates antitrypsin deficiency. A methionyl-seryl group (positions 358 and 359) is present at the point where the inhibitor binds to the active site of elastase. The smoke promotes the oxidation of Methionine residues to the corresponding sulfoxide. This results in the inactivation of the protein, which fails to bind elastase. The active elastase then destroys the lung tissue.
Treatment
Treatment involves replacing the missing AAT protein. The protein derived from donated human plasma is given intravenously each week. This augmentation therapy is thought to arrest the course of the disease and halt any further damage to the lungs. Long-term studies on the effectiveness of A1AT replacement therapy are not available. It is currently recommended that patients begin augmentation therapy only after the onset of emphysema symptoms.
Quitting smoking is crucial.
Other treatments specifically for emphysema and cirrhosis are also used.
Treatments currently being studied include recombinant and inhaled forms of A1AT. Antioxidants are also supplemented.