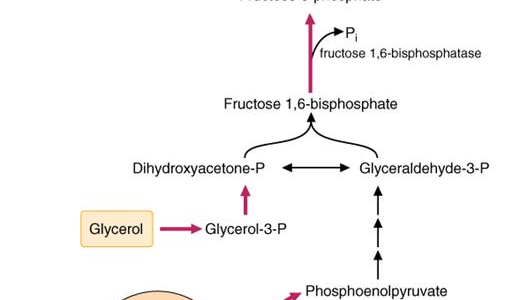
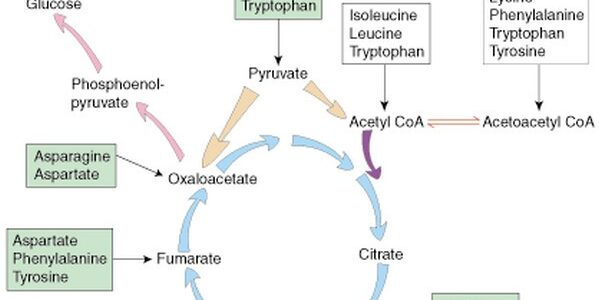
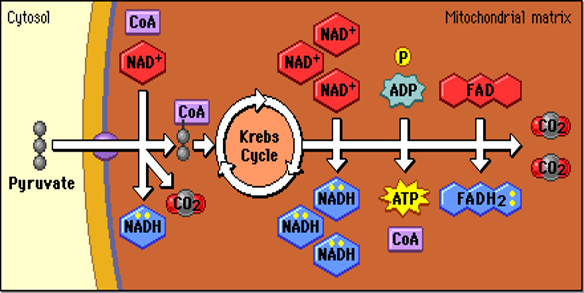
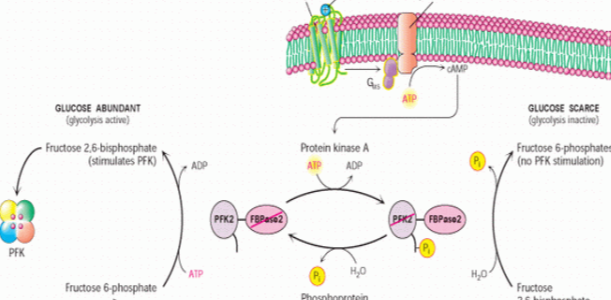
Quick Revision Series
Glycogen Storage Diseases- Multiple-Choice Questions
Posted in:
Energy metabolism ,
Learning resources ,
Library ,
Metabolism of Carbohydrates ,
Multiple-choice questions ,
Multiple-choice questions ,
Multiple-Choice questions ,
Practice questions ,
Practice questions ,
Question Bank ,
Question Bank ,
Quick Revision Series ,
USMLE Content ,
USMLE Style questions ,
USMLE styled question bank ,